Isomorphic Lab’s New Drug Design Engine Promises to Outdo AlphaFold 3

Isomorphic Labs is giving researchers a glimpse of its latest AI-driven drug discovery platform: the Isomorphic Labs Drug Design Engine (IsoDDE) promising significant improvements in overall prediction capabilities. Being positioned as a step beyond AlphaFold-style structure prediction, the system aims to unify multiple predictive tasks—structure, binding, affinity, and pocket discovery—into a single computational framework.
For readers already familiar with structure prediction, the announcement represents an important conceptual shift: from “predicting what molecules look like” to “designing medicines end-to-end in silico.”
AlphaFold and its successors fundamentally changed structural biology by predicting protein shapes from amino acid sequences. But accurate structure alone does not create drugs. Drug discovery requires at least four interconnected capabilities:
- Predicting biomolecular structure
- Understanding how molecules bind
- Estimating binding strength (affinity)
- Discovering new binding pockets
Isomorphic Labs argues that earlier models excelled primarily at predicting biomolecular structure, while real drug design requires integrated predictions across all four. IsoDDE is described as a unified computational drug-design system that bridges this gap by combining multiple predictive tasks into one framework.
Generalisation: The Central Technical Challenge
One of the most important claims in the announcement is not just accuracy—but generalisation to unseen biology. Many deep-learning models perform well on systems similar to their training data. But drug discovery frequently targets:
- Novel protein conformations
- Uncharacterised pockets
- Previously unseen chemical scaffolds
Benchmarks cited by Isomorphic Labs indicate that IsoDDE more than doubles AlphaFold 3’s accuracy on difficult protein-ligand cases that are highly dissimilar to training data. This is significant because most real-world drug targets lie precisely in these unexplored regions of chemical and biological space.
Generalisation refers to a model’s ability to make correct predictions on data it has never seen before.
- Poor generalisation: works only on familiar proteins or ligands
- Strong generalisation: works on new diseases, new targets, and novel chemistries
In drug discovery, generalisation is critical because every new therapeutic target is, by definition, an unseen system.
Modelling Induced Fit and Cryptic Pockets
The report highlights the model’s ability to capture complex structural phenomena, including:
- Induced fit: when a protein changes shape to accommodate a ligand
- Cryptic pockets: binding sites that only appear when a ligand is present
IsoDDE reportedly predicts these out-of-distribution events accurately—even when they are far from training data examples.
Induced fit : Proteins are not rigid objects. When a drug molecule approaches, the protein may change shape to allow binding. Predicting this dynamic change is difficult because it requires modelling flexible structures and the bound state may not exist in the training data
Cryptic pockets: These are hidden binding sites that do not appear in the unbound protein structure and only form when a ligand interacts with the protein. Many “undruggable” targets become druggable if cryptic pockets are discovered.
Expanding to Complex Biologics
While small-molecule drugs remain dominant, biologics—especially antibodies—are increasingly important.
IsoDDE reportedly:
- Outperforms AlphaFold 3 by 2.3× on antibody–antigen predictions
- Outperforms another model (Boltz-2) by 19.8× in high-accuracy regimes
The system shows strong performance on the CDR-H3 loop, the most variable and difficult antibody region to predict.
What Is the CDR-H3 Loop?
Antibodies bind targets through small flexible regions called Complementarity-Determining Regions (CDRs). There are six CDRs in an antibody where CDR-H3 is the most variable and structurally complex. It often determines the binding specificity, binding strength and therapeutic effectiveness
Accurate prediction of CDR-H3 geometry is one of the hardest problems in antibody modelling.
Binding Affinity: The Core of Drug Optimisation
Knowing the structure of a protein-ligand complex is only the first step. Drug design depends on predicting binding affinity—how strongly the drug attaches to the target.
IsoDDE claims:
- Performance exceeding all deep-learning methods on several benchmarks
- Accuracy surpassing physics-based approaches such as FEP (Free Energy Perturbation)
- Much lower computational cost and runtime
Binding affinity is a quantitative measure of how strongly a drug binds its target.
- High affinity → strong binding → potentially more effective drug
- Low affinity → weak binding → less effective
Free Energy Perturbation (FEP) is a physics-based simulation technique that:
- Uses molecular dynamics
- Computes the change in free energy when a molecule is modified
- Is considered a “gold standard” for affinity prediction
However, FEP Requires large computational resources that often depends on experimental structures which take time and money to visualize. IsoDDE claims to match or exceed FEP accuracy without these constraints.

Blind Pocket Discovery: Expanding the Druggable Proteome
One of the most intriguing capabilities described is ligand-independent pocket detection. IsoDDE claims to be able to identify potential binding pockets using only protein sequence, discover novel sites far from training data and achieve performance approaching experimental fragment-soaking techniques. All this has to be verified by independent researchers.
A cited example involves cereblon, where a novel allosteric cryptic pocket was experimentally discovered after years of study. IsoDDE reportedly predicted both: The known binding pocket as well as the newly discovered cryptic site — and did so using only the sequence as input.
Toward a Unified Drug Design Engine
Conceptually, IsoDDE represents a move from single-task structural AI to a multi-objective molecular reasoning system.
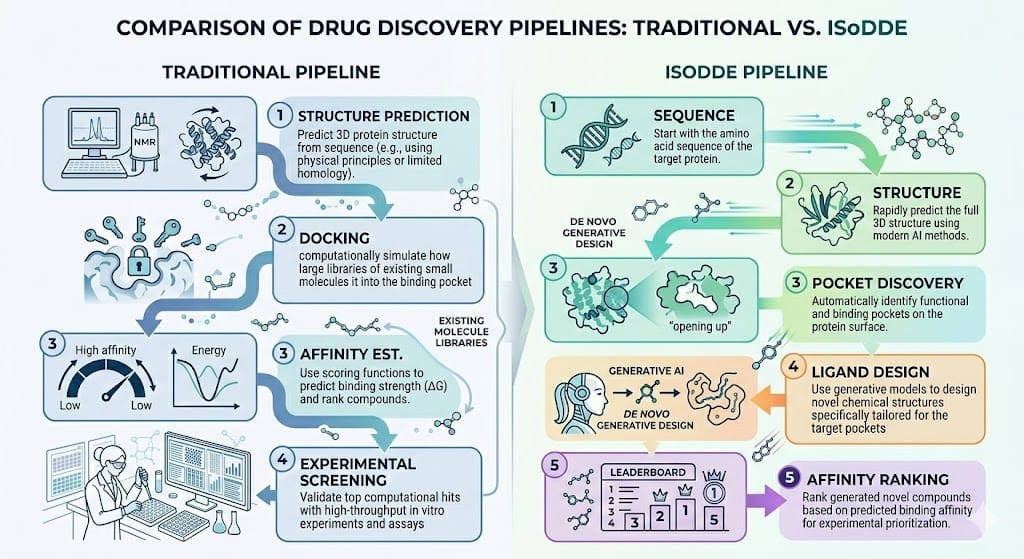
All within one integrated computational system.
This architecture suggests a shift from: “AI as a structural tool” -> “AI as the primary drug design engine”
Strategic and Scientific Significance
If the reported capabilities hold across real programs, several implications follow:
1. Shorter discovery cycles: Accurate affinity prediction and pocket discovery could reduce the need for iterative synthesis cycle and accelerate hit-to-lead and lead optimisation.
2. Expansion of target space: Cryptic pocket detection may unlock previously undruggable proteins and enable allosteric and degradation-based therapies.
3. Shift toward fully in-silico design: The long-term goal is clear, to design candidate drugs computationally before synthesis, rather than relying on large-scale screening.
Key Technical Claims (Summary)
According to the announcement, IsoDDE more than doubles AlphaFold 3 accuracy on difficult protein-ligand benchmarks and can potentially outperforms competitors on antibody–antigen prediction. While also surpassing physics-based methods in binding affinity prediction and discovers cryptic pockets using sequence alone. More importantly it integrates multiple drug-design tasks into one system leading to autonomous drug design.
| Phase 1: Structural prediction | Phase 2: Interaction modelling | Phase 3: Autonomous design engines |
| AlphaFold-style models Predict protein structures | Protein-ligand complexes Antibody interfaces Binding affinity | Discover pockets Generate molecules Rank candidates Optimise in silico |
IsoDDE is positioned as a transition from Phase 2 to Phase 3 if validated in large-scale clinical pipelines, such engines could shift pharmaceutical R&D from: Empirical, lab-first discovery to Predictive, computation-first medicine design.