Thousands of Single Cell Transcriptomes Analyzed in Parallel
For a long time, gene expression was measured in samples of thousands of cells. The results were an average of the whole cell population. But now, genomic technology makes possible RNA sequencing (RNA-seq) in single cells. Experiments in cell differentiation, responses to stimulation, tracking of heterogeneous cell sub-populations or characterization of cell identity and function have all happened thanks to single cell transcriptomics, where hundreds of cells are individually analyzed. This has a profound impact in the study of cancer, tissue regeneration and other fields. Despite the advance, hundreds of cells are sometimes not sufficient. For experiments that demand statistical power or finding a very rare cell, scientists need a new technique that allows analyzing the transcriptome of an unlimited amount of cells.
Drop-seq and inDrop: two very similar techniques
Two laboratories from Harvard University have independently developed two techniques that allow scaling up single cell transcriptomics. Dr. Kirschner’s and Dr. McCarroll found very similar solutions: they used microfluidic tubes, water and oil to create thousands of water droplets, each containing a set of barcoded primers and a cell. The barcodes allow identifying from which cell the transcripts come. All transcripts from all cells can be reverse transcribed and sequenced in parallel.
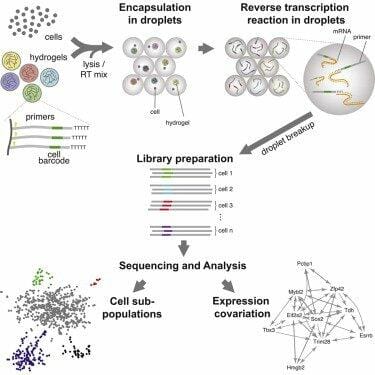
inDrop method. From: http://www.cell.com/cell/abstract/S0092-8674%2815%2900500-0
With his Drop-seq method, Steven McCarroll and his team analyzed 44,808 mouse retina cells and identified 39 transcriptionally different cell populations. Dr. Kirschner’s team has used its inDrop technology to analyze mouse embryonic stem cells, and discovered the cell population structure and their gene regulatory linkages.
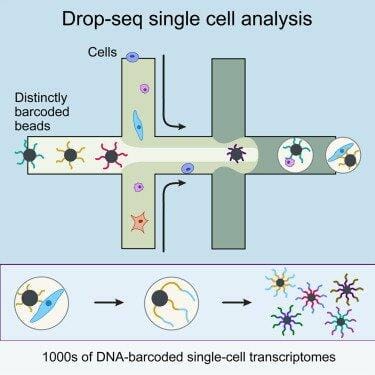
Drop-seq method. From: http://www.cell.com/abstract/S0092-8674%2815%2900549-8
Both works have been published in the journal Cell. The possible applications of both techniques are enormous, as massive parallel single cell transcriptomics will be progressively implanted as a routine technique in every laboratory.
Source: TheScientist