The Day Biology Changed Forever: Why April 14th Matters in the Story of the Human Genome

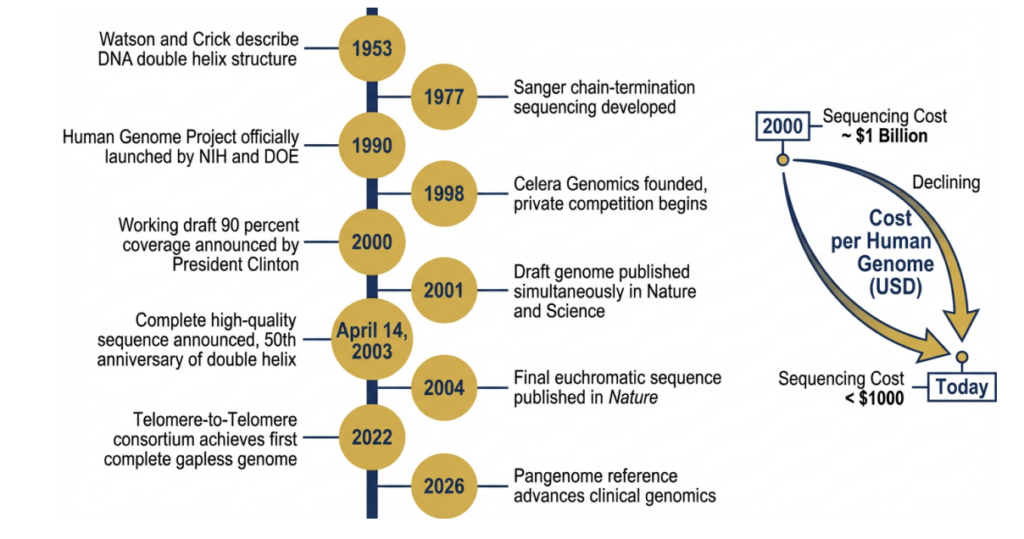
Sixteen years ago today on April 14, 2003, one of the most ambitious scientific endeavors in history reached its defining milestone. The Human Genome Project—a 13-year international effort to map and understand all the genes of human DNA—was declared complete. Timed deliberately to coincide with the 50th anniversary of the discovery of the DNA double helix, this date marked not just the end of a project, but the beginning of a new era in biology, medicine, and data-driven science.

of the DNA double helix in 1953 to the triumphant completion announcement on April 14,
2003, and the transformative applications that followed. The project catalysed a 3 → $0.001
billion-per-genome cost revolution and spawned the era of precision medicine.
When the Human Genome Project (HGP) began in 1990, sequencing the entire human genome seemed almost impossibly complex and unnecessary. The human genome contains roughly 3 billion base pairs—chemical letters that encode the instructions for building and maintaining a human being. The goal was not only to sequence these letters but to make them accessible for researchers worldwide and help decode what every nucleotide’s function.
Led by institutions like the National Institutes of Health and involving scientists across the United States, Europe, and Asia, the project also ran parallel to private efforts spearheaded by Craig Venter and the company Celera Genomics. Meanwhile, public leadership figures such as Francis Collins played a pivotal role in ensuring the genome would remain a shared scientific resource.
By April 14, 2003, the project had successfully sequenced and stitiched over 99% of the human genome with remarkable accuracy—far exceeding initial expectations. Over thirteen years, researchers across twenty sequencing centres in six nations decoded the approximately 3.2 billion base pairs of the human genome at an accuracy surpassing one error per 10,000 bases. The road was paved with formidable technical obstacles: the intractable repetitiveness of the human genome, the computational complexity of assembling billions of short reads, the engineering demands of scaling Sanger capillary sequencing to industrial throughput, and the political and organisational challenges of coordinating a global consortium under fierce competitive pressure. This article traces the arc of that journey — its methodological battles, the singular importance of April 14th as a date in scientific history, and the way the HGP’s legacy continues to reverberate through genomics, medicine, and biotechnology twenty-three years on.
Why April 14th Is More Than a Date
The significance of April 14th lies in its symbolism as much as its scientific achievement. It represents:
1. The Transition from Discovery to Application
By scheduling the completion announcement for the 50th anniversary of that paper that announced the discovery of DNA (celebrated on April 25, but the project completion was declared April 14), the organisers drew a line of continuity from the first glimpse of DNA’s architecture to the reading of its full three-billion-letter text. Before this milestone, biology was largely descriptive. After it, biology became predictive and computational. Researchers could now identify genes associated with diseases, understand genetic variation, and begin to design targeted therapies.
2. The Birth of Genomic Medicine
The completion of the genome laid the foundation for personalized medicine. Today, treatments for cancers, rare diseases, and genetic disorders often depend on genomic insights that trace directly back to the HGP.
3. The Democratization of Biological Data
One of the most important decisions made during the HGP was to make the data publicly available. This open-access model accelerated innovation globally, enabling startups, academic labs, and pharmaceutical companies to build upon a shared foundation.
4. A New Relationship Between Biology and Technology
The HGP catalyzed advances in sequencing technologies, computational biology, and bioinformatics. What once cost billions of dollars and took over a decade can now be done in hours for a fraction of the cost.
The April 14, 2003 declaration was not a sudden surprise. The world had gotten its first taste of the genome in June 2000, when President Clinton and UK Prime Minister Tony Blair jointly announced the completion of a working draft — a mosaic covering roughly 90% of the human genome’s gene-encoding regions. That moment had its own gravitas, shared between the public consortium led by Francis Collins at the National Institutes of Health and Craig Venter’s upstart private company, Celera Genomics. But the April 2003 announcement was the “finished” sequence: a rigorous, high-quality reference with greater than 99.99% accuracy. It was the document that science had been promised.
Forging The Blueprint: Background & Origins
The idea of sequencing the entire human genome was first floated seriously in the mid-1980s, and when it was, many prominent scientists considered it outright lunacy. The genome is enormous — roughly 3.2 billion nucleotide base pairs spread across 23 pairs of chromosomes — and sequencing even a few thousand bases in the early 1980s was a hard day’s work in a specialised laboratory. The Sanger chain-termination method, developed by Frederick Sanger and colleagues in 1977 and ultimately earning Sanger his second Nobel Prize, had made sequencing reliable, but not fast [Sanger et al., 1977].
The formal launch of the Human Genome Project came on October 1, 1990, jointly announced by the National Institutes of Health and the Department of Energy in the United States, with partners in the United Kingdom, France, Germany, Japan, and China soon joining under the banner of the International Human Genome Sequencing Consortium (IHGSC). The project’s initial budget was estimated at $3 billion over 15 years — an unprecedented public investment in basic science, and one that attracted immediate scrutiny over whether such a resource commitment was justified.
Two philosophical pillars guided the project’s design. The first was a commitment to making all data freely and immediately available — enshrined in the Bermuda Principles of 1996, which mandated that all sequence data be deposited in public databases (primarily GenBank) within 24 hours of generation [Collins et al., 1998]. The second was a hierarchical approach to sequencing: rather than attempting to read the genome randomly from end to end, researchers would first map it physically, break it into manageable chunks called bacterial artificial chromosomes (BACs), sequence each chunk individually, and then assemble the results by overlapping known clone boundaries.
This clone-by-clone strategy was methodologically conservative but scientifically sound. Each BAC insert spanned roughly 150,000 to 200,000 bp, a size amenable to complete and accurate assembly. By anchoring BACs to physical maps of each chromosome, the consortium could be confident about where every piece of sequenced DNA belonged in the genome’s grand architecture.
Technical Challenges to Achieve the Impossible
Calling the HGP merely a scientific achievement undersells the magnitude of its engineering. The project required building new instruments, new software, new data infrastructure, and new organisational frameworks, often simultaneously and always under enormous time pressure.
The Repetition Problem: When the Genome Defies Assembly
Perhaps the single greatest biological obstacle the HGP faced was the sheer repetitiveness of the human genome. Approximately half of all human DNA consists of repetitive elements — short tandem repeats (STRs), satellite sequences, and transposable elements such as LINEs (Long Interspersed Nuclear Elements) and SINEs (Short Interspersed Nuclear Elements). More treacherous still were segmental duplications: stretches of DNA longer than 15 kilobases that are more than 97% identical to another region somewhere else in the genome [International Human Genome Sequencing Consortium, 2001].
The hierarchical clone-by-clone strategy was specifically designed to mitigate the repetition problem. Because each BAC clone was mapped to a unique chromosomal position before sequencing, the assembler knew a priori which chromosome and which locus a read came from. This localisation dramatically reduced the ambiguity introduced by repetitive elements, though it did not eliminate it entirely. Regions of the genome where repetitive elements clustered densely — centromeres, telomeres, and the short arms of acrocentric chromosomes — remained unresolvable even in the 2003 finished sequence.
Scaling Sanger Sequencing to Industrial Throughput
The Sanger method, in its original form, was a manual procedure. A researcher would set up a sequencing reaction in a test tube, separate the resulting chain-terminated fragments by gel electrophoresis, and read the sequence from the gel pattern, base by base, by eye. At peak performance, a skilled technician might read 500 bases per reaction on a good day. Scaling this to a genome of 3.2 billion bases required an almost total reimagining of the process.
The key breakthrough came with capillary electrophoresis, which replaced flat slab gels with a bundle of 96 or 384 narrow glass capillaries, each running a separate sequencing reaction simultaneously. Instruments like the ABI 3700 and its successor the ABI 3730xl automated the loading, separation, fluorescence detection, and base-calling process into a single robotic workflow. A single machine could generate roughly 1,000 sequencing reads per day, each up to 800 bases long, with minimal human intervention.
Key Fact: At its peak in 2001–2003, the HGP was producing genome sequence at a rate that would have seemed impossible a decade earlier. The Wellcome Trust Sanger Institute alone operated hundreds of ABI 3700 sequencing machines running 24 hours a day, seven days a week, as part of a global effort spanning the United States, the United Kingdom, France, Germany, Japan, and China.
Even so, the throughput of capillary sequencing was insufficient to sequence the genome from scratch at the required depth. Researchers needed to sequence every position of the genome multiple times — at least 8–10× “coverage” — to distinguish genuine sequence variants from sequencing errors. This meant generating many tens of billions of raw base pairs, which had to be stored, backed up, quality-controlled, and transmitted between centres in real time.
Computational Bottlenecks: Assembling the Puzzle
The computational demands of the HGP were staggering for their era. Assembly — the process of stitching together millions of overlapping short reads into a coherent linear sequence — required algorithms capable of comparing every read against every other read, identifying overlaps, resolving conflicts, and building a consensus sequence, all while tracking provenance information so that errors could be detected and corrected.
The PHRED/PHRAP/CONSED software suite, developed at the University of Washington, became the workhorse of the public consortium [International Human Genome Sequencing Consortium, 2001]. PHRED assigned quality scores to each base call; PHRAP used those scores to assemble overlapping reads into “contigs”; and CONSED provided an interactive environment for human “finishers” to manually inspect and correct problematic regions. This pipeline was designed for clone-by-clone work, where the input to the assembler was a manageable set of reads from a single BAC rather than the entire genome at once.
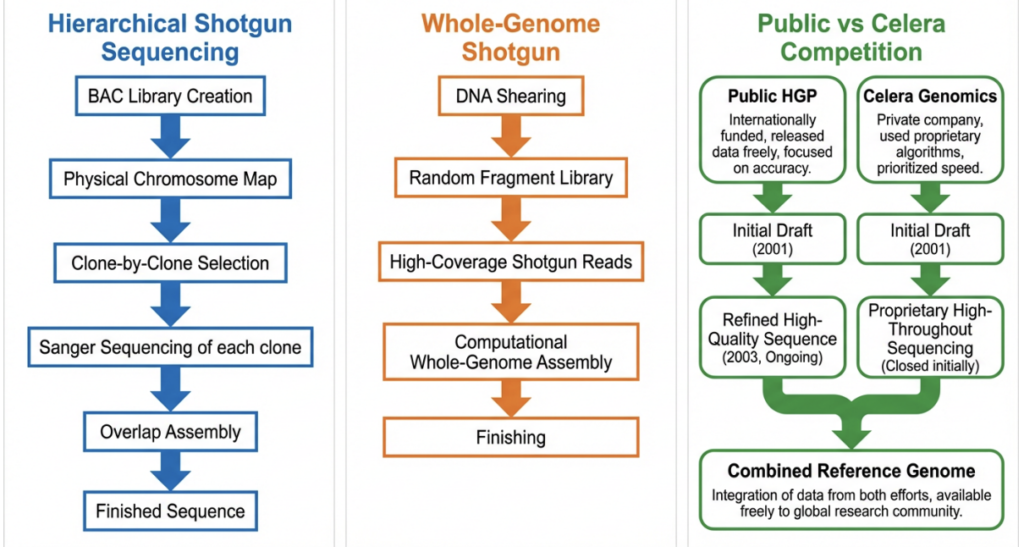
The competitive pressure exerted by Celera Genomics forced the public consortium to also develop whole-genome shotgun (WGS) capabilities [Weber and Myers, 1997]. In WGS — the strategy that Celera championed — the entire genome is sheared randomly into small fragments, both ends of each fragment are sequenced (producing “paired-end reads”), and the resulting reads are assembled computationally without reference to a pre-existing physical map. The advantage of WGS is speed: by parallelising all the sequencing at once, Celera was able to generate the equivalent of a draft human genome sequence far faster than the public consortium had anticipated [Venter et al., 2001].
The disadvantage of WGS, as the public consortium’s computational biologists had argued years earlier, was its vulnerability to repetitive sequences [Green, 1997]. Without the anchor of a physical map, a whole-genome assembler cannot distinguish between reads from two nearly identical segmental duplications. The Celera assembler — built around the Celera Assembler software — partially addressed this by incorporating public consortium data as a scaffold, effectively combining the best of both worlds [Venter et al., 2001]. Even so, analyses published after the HGP’s completion confirmed that WGS assemblies of that era systematically underrepresented or misassembled large segmental duplications, leaving gaps that would not be closed for nearly two more decades [International Human Genome Sequencing Consortium, 2004].
Data Management at Unprecedented Scale
Each day, sequencing centres around the world were generating terabytes of raw sequence data — an astonishing volume for early-2000s computing infrastructure. The data had to be transmitted over the then-nascent internet, deposited into GenBank, and made publicly accessible within 24 hours under the Bermuda Principles. This required dedicated high-speed network links between sequencing centres, custom software for automated data submission, and database infrastructure at the National Center for Biotechnology Information (NCBI) capable of ingesting and serving data at rates that stressed the existing architecture.
Visualisation tools were also essential to get an overall picture of the progress made. The UCSC Genome Browser, developed by Jim Kent and David Haussler’s group, provided the scientific community with a web interface to explore the accumulating sequence data, overlay functional annotations, and compare human sequence with other organisms [Kent et al., 2002]. Kent wrote the assembly code for the human genome — some 10,000 lines of C — in just four weeks, a heroic feat of software engineering that compressed a read-overlap-graph algorithm to run within the memory constraints of early 2000s hardware.
International Coordination: The Bermuda Principles and Beyond Running a 13-year, $3 billion scientific project across six countries, twenty sequencing centres, and thousands of researchers required more than good science. It required governance. The IHGSC adopted the Bermuda Principles to prevent data being locked behind proprietary walls, and it divided the sequencing labour geographically: specific chromosomes were assigned to specific centres (for example, the Wellcome Trust Sanger Institute in the UK took primary responsibility for sequencing chromosomes 1, 6, 9, 10, 13, 20, 22, and the X chromosome, and contributed roughly one-third of the finished human genome sequence).
The entry of Celera Genomics in 1998 transformed the project’s politics. Craig Venter, a former NIH scientist with a history of iconoclastic moves in genomics, announced that his newly founded company would sequence the genome faster and cheaper than the public consortium, and would make the data available — under restrictive licensing terms that the public consortium found unacceptable. The competitive race that ensued, while often publicly acrimonious, was scientifically productive: fear of being scooped accelerated the public consortium’s timeline by roughly two years [Collins et al., 2003].
Long Road Still Left Unfinished
Despite the triumphant declaration of April 14, 2003, specialists in genomics were well aware that the finished sequence was not, in fact, complete. The regions excluded from the finished assembly — centromeres, telomeres, the acrocentric chromosome short arms, and regions of dense heterochromatin — account for approximately 8% of the total genome and contain hundreds of thousands of repetitive base pairs that Sanger-based sequencing, even with the hierarchical clone approach, could not resolve. These were not trivial gaps; centromeres and telomeres play critical roles in chromosome segregation during cell division, and their disruption underlies certain chromosomal instability syndromes.
The resolution of these regions had to wait until the advent of long-read sequencing technologies: PacBio’s HiFi sequencing and Oxford Nanopore’s ultra-long reads, which can span entire repetitive arrays in a single read, eliminating the ambiguity that had defeated earlier approaches. In 2022, the Telomere-to-Telomere (T2T) Consortium published the first truly complete sequence of a human genome — every base pair, including the previously inaccessible centromeric and telomeric regions, resolved for the first time [Nurk et al., 2022]
The T2T-CHM13 assembly published in 2022 added approximately 200 million base pairs to the reference human genome — sequence that had never been read in the 50-year history of genomics. It revealed approximately 2,000 previously unknown genes and corrected thousands of errors in the GRCh38 reference that had been propagated through a generation of genomic studies [Nurk et al., 2022].
The Ripple Effects Still Expanding
The true significance of April 14, 2003, becomes clearer with each passing year. The HGP reference genome serves as the foundation upon which successive technological and scientific layers have been built — from next-generation and long-read sequencing platforms, through population-scale pangenome references, to the apex of individualised precision medicine. Entire fields—genomics, transcriptomics, proteomics—owe their acceleration to this milestone. Projects like the Human Cell Atlas, large-scale biobanks, and AI-driven drug discovery platforms all trace their lineage back to the HGP.
Moreover, companies working in CRISPR gene editing, synthetic biology, and precision diagnostics are fundamentally operating on the blueprint first completed on that day.
Not an Ending, but a Beginning
It’s tempting to view April 14th as the finish line of a grand scientific race. In reality, it was the starting point of a far more complex journey. Sequencing the genome told us what is there—but understanding how it works, how it varies, and how to manipulate it safely remains an ongoing challenge.
Even today, scientists continue refining the genome. In 2022, the Telomere-to-Telomere (T2T) consortium filled in previously missing regions, showing that the “complete” genome of 2003 was, in fact, a highly accurate draft.
A Legacy That Continues to Shape the Future
April 14th stands as a landmark not just for genomics, but for how humanity approaches complex problems. It demonstrated the power of international collaboration, open science, and long-term vision.
In many ways, the Human Genome Project did for biology what the internet did for communication—it created a foundational layer upon which everything else could be built.
And more than two decades later, we are still only beginning to understand its full impact.
A Look At Some of The Faces Behind the Human Genome Project

Francis Collins was the public face and scientific leader of the Human Genome Project. As director of the National Human Genome Research Institute, he championed open-access data sharing and coordinated global collaboration. A physician-geneticist, Collins later went on to serve as Director of the National Institutes of Health, shaping biomedical research policy for over a decade.

Craig Venter led the private-sector effort through Celera Genomics, pushing a faster sequencing strategy using whole-genome shotgun methods. His competitive approach accelerated the overall timeline of the project. Venter is widely regarded as a pioneer of modern genomics and synthetic biology.
James Watson, best known as co-discoverer of the DNA double helix, he was an early advocate of the Human Genome Project. He helped set its initial direction but stepped down in 1992. His early advocacy was crucial in securing support for large-scale genome sequencing.

Eric Lander played a central role in the computational and analytical side of the Human Genome Project. As a leader at the Broad Institute, he contributed to genome mapping and interpretation, helping translate raw sequence data into meaningful biological insights.

John Sulston, a British biologist and Nobel laureate, led the UK’s contribution to the Human Genome Project at the Sanger Institute. He was a strong advocate for keeping genomic data in the public domain and opposed efforts to privatise genetic information.

Conclusion or the Beginning
On April 14, 2003, humanity received the most complex message it had ever read: the 3.2-billion letter instruction manual for building a human being. The Human Genome Project that delivered it was a triumph of international science, computational innovation, engineering ingenuity, and political will operating simultaneously across six nations and two decades. Its completion was declared on a date chosen to echo the 50th anniversary of the double helix — a conscious linking of the structural beginning of molecular biology to its informational culmination.
The technical challenges the project overcame — repetitive sequences that defeated naive assembly, sequencing throughput that had to be scaled by orders of magnitude, computational infrastructure that had to be invented alongside the science, and international coordination that had to be maintained against the fierce competition of the private sector — were each individually daunting.
Together, they constituted perhaps the greatest sustained technical challenge that biology has ever imposed upon itself. That the consortium met them, and met them on schedule, remains a source of justified pride for everyone who contributed. Twenty-three years later, the HGP’s most lasting legacy is not the sequence file stored in GenBank. It is the culture of open science that the Bermuda Principles enshrined, the computational biology it catalysed, the genomics industry it spawned, the clinical genomics it enabled, and the public understanding of genetics it transformed. The reference genome published in April 2003 has since been superseded, extended, and enriched — first by the final euchromatic sequence of 2004, then by the T2T-CHM13 gapless assembly of 2022, and now by the emerging Human Pangenome Reference. Each iteration makes the original book richer, more accurate, and more representative of all of humanity.
April 14 is not just a date in scientific history. It is a reminder that when the world’s best scientists set aside competition and commit to open, rigorous, cumulative knowledge-building — when they agree, as the Bermuda Principles demanded, that the genome belongs to all of humanity — the results can be transformative beyond any individual career, institution, or generation. The code of life was cracked on April 14, 2003. The reading of what it means is still underway.
References
- Francis S. Collins, Aristides Patrinos, Elke Jordan, Aravinda Chakravarti, Ray Gesteland, LeRoy Walters, the members of the DOE, and NIH planning groups. New goals for the U.S. human genome project: 1998–2003. Science, 282(5389):682–689, 1998. doi: 10.1126/science.282.5389.682.
- Francis S. Collins, Michael Morgan, and Aristides Patrinos. The human genome project: Lessons from large-scale biology. Science, 300(5617):286–290, 2003. doi: 10.1126/science.1084564.
- Eric D. Green, Chris Gunter, Leslie G. Biesecker, Valentina Di Francesco, Christine L. Easter, Elise A. Feingold, Adam L. Felsenfeld, David J. Kaufman, Elaine A. Ostrander, William J. Pavan, et al. Strategic vision for improving human health at The Forefront of Genomics. Nature, 586:683–692, 2020. doi: 10.1038/s41586-020-2817-4.
- Philip Green. Against a whole-genome shotgun. Genome Research, 7(5):410–417, 1997. doi: 10.1101/gr.7.5.410.
- Margaret A. Hamburg and Francis S. Collins. The path to personalized medicine. New England Journal of Medicine, 363(4):301–304, 2010. doi: 10.1056/NEJMp1006304.
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature, 409:860–921, 2001. doi: 10.1038/35057062.
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature, 431:931–945, 2004. doi: 10.1038/nature03001.
- W. James Kent, Charles W. Sugnet, Terrence S. Furey, Krishna M. Roskin, Tom H. Pringle, Alan M. Zahler, and David Haussler. The human genome browser at UCSC. Genome Research,
12(6):996–1006, 2002. doi: 10.1101/gr.229102. - Wen-Wei Liao, Mobin Asri, Jana Ebler, Daniel Doerr, Marina Haukness, Glenn Hickey, Shuangjia Lu, Julian K. Lucas, Jean Monlong, Hannah J. Abel, et al. A draft human pangenome reference.
Nature, 617:312–324, 2023. doi: 10.1038/s41586-023-05896-x. - Elaine R. Mardis. A decade’s perspective on DNA sequencing technology. Nature, 470:198–203, 2011 doi: 10.1038/nature09796.
- Sergey Nurk, Sergey Koren, Arang Rhie, Mikko Rautiainen, Andrey V. Bzikadze, Alla Mikheenko, Mitchell R. Vollger, Nicolas Altemose, Lev Uralsky, Ariel Gershman, et al. The complete sequence of a human genome. Science, 376(6588):44–53, 2022. doi: 10.1126/science.abj6987.
- Frederick Sanger, Steven Nicklen, and Alan R. Coulson. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences, 74(12):5463–5467, 1977. doi: 10.1073/pnas.74.12.5463.
- J. Craig Venter, Mark D. Adams, Eugene W. Myers, Peter W. Li, Richard J. Mural, et al. The sequence of the human genome. Science, 291(5507):1304–1351, 2001. doi: 10.1126/science. 1058040.
- James D. Watson and Francis H. C. Crick. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature, 171:737–738, 1953. doi: 10.1038/171737a0.
- James L. Weber and Eugene W. Myers. Human whole-genome shotgun sequencing. Genome Research, 7(5):401–409, 1997. doi: 10.1101/gr.7.5.401.