New Method Predicts Protein Motion from Static Structures
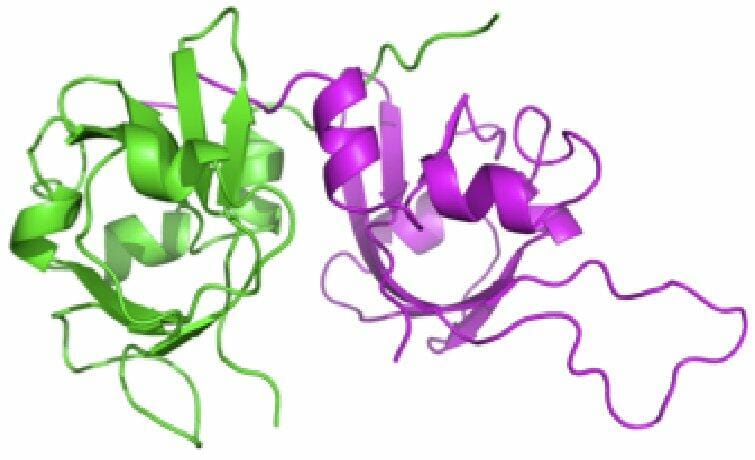
Researchers from Oregon Science and Health University have gone further in the understanding of protein motion. The study “Parsimony in Protein Conformational Change” has been published in Structure and contains advances in how proteins move to perform their functions. This discovery can have an impact in the understanding of certain diseases at a molecular level and in the design of drugs to treat them.
Protein structures are mainly solved by X-Ray Crystallography. This is a powerful technique but yields static pictures of the protein, crystallized in different conformations. Nuclear Magnetic Resonance Spectroscopy allows to obtain dynamic data of proteins in solution, but this technique has important limitations in protein size. As a result, less than one percent of the protein structures are solved by this method.
Michael Chapman and coworkers have found a way to tell movement from the static structures obtained by X-Ray Crystallography. They designed a software that, from two static structures of the same protein, predicts hot-spots of flexibility or hinge points. The team has proved the theory that stated that proteins use the most efficient motion ranges, and will help in understanding protein interactions and their cellular function.
Source: http://www.ohsu.edu/xd/about/news_events/news/2015/06-18-researchers-bring-to-lif.cfm
Reachout to Researchers with Our Extensive Marketing Network
Modern scientific marketing partner built for life science brands