Genome Editing Dampens Mitochondrial DNA Mutation Heritability
Researchers from Salk Institute in San Diego, CA have presented a breakthrough study on the elimination of defective mitochondrial DNA (mtDNA). The use of genome editing tools -restriction endonucleases and transcription activator-like effector nucleases (TALENs)- allowed them to succesfully target specific mtDNA mutations and reduce their transgenerational tranmission. The tests have been carried out on mice oocytes and one cell embryos. The findings could be applied in the future to treat human mitochondrial diseases.
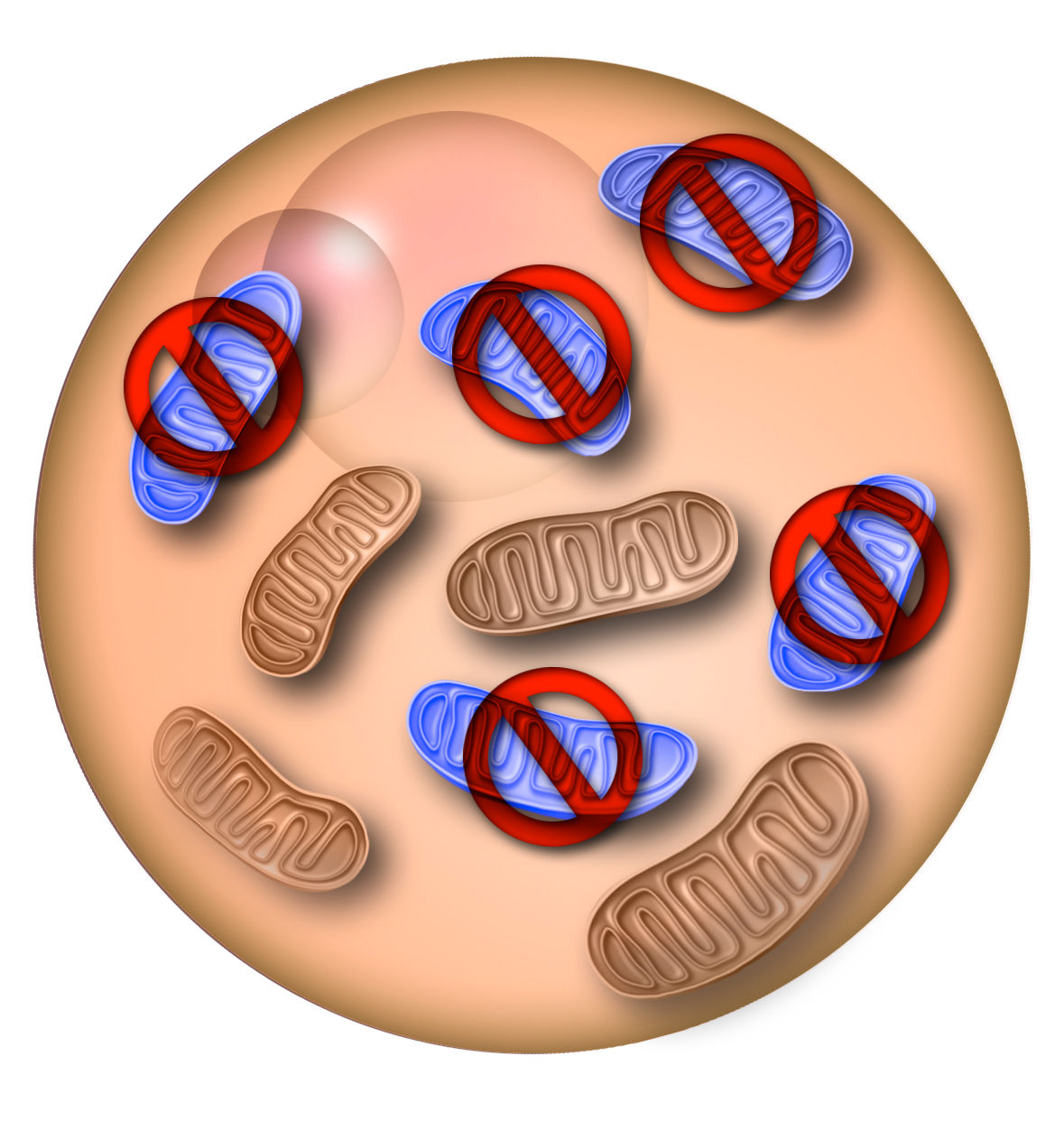
Mitochondria are fundamental for the cell, being energy production their main duty. Defects in these organelles are especially dramatic for high energy-consuming organs like the heart and the brain. There is no cure for mitochondrial diseases, but their transmission can be reduced by pre-impantation genetic diagnosis; however, the risk in far from zero, and embryo manipulation can affect its viability. A recently developed alternative is to transfer the genome into a donor oocyte or embryo that provides the healthy mitochondria, but the bioethical implications of introducing DNA from a third person reduce this technique’s appeal.
Recent studies accomplished the reduction of defective mtDNA in human somatic cells and mature mice to levels that effectively eliminated the sick phenotype. But the group led by Izpisua Belmonte managed, for the first time, to target and eliminate a specific mtDNA mutation in the germline and thus reduce transgenerational transmission.
Transgenerational transmission below phenotypic threshold
First, Reddy et al. used NZB/BALB heteroplasmic mice as a proof of principle. These rodents have two mtDNA haplotypes, BALB and NZB, that simulated the mutated and healthy mtDNA coexisting in most mitochondria disease patients. The researchers prevented the germline transmission of either haplotype by using mitochondria-targeted restriction endonucleases or TALENs, a kind of nucleases engineered to target and cut specific DNA sequences.
Next, they fused human patient cells containing mutated mtDNA with mice oocytes. By using the same principle, the Salk Institute team was able to reduce transgenerational transmission of human mutated mtDNA responsible for Leber’s hereditary optic neuropathy (LHOND), and neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). Not being able to eliminate all the mutated copies was not a concern, as long as the percentage is below the threshold levels (60%–95%) required for clinical defects to manifest in the next generation.
In short, Izpisua Belmonte’s team and collaborators have developed a very promising technique for the future treatment of human mitochondrial diseases.
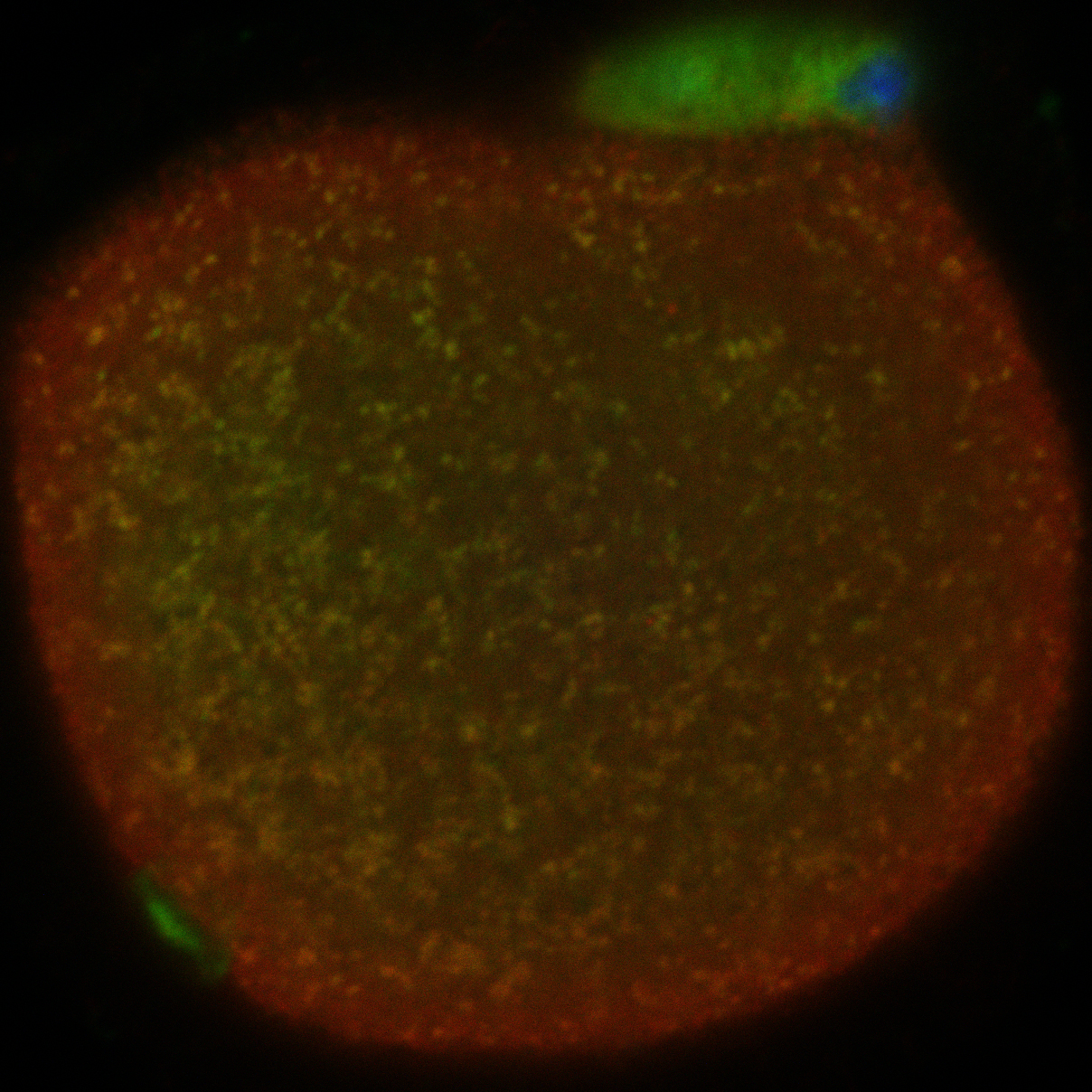
Live cell imaging allows observing the oocyte; yellow dots indicate localization of nucleases with mitochondria.
Sources:
http://www.cell.com/cell/fulltext/S0092-8674%2815%2900371-2
http://www.salk.ed/news/pressrelease_details.php?press_id=2077
Reachout to Researchers with Our Extensive Marketing Network
Modern scientific marketing partner built for life science brands