Protein Dynamics Are Predictable from The Amino Acid Sequence
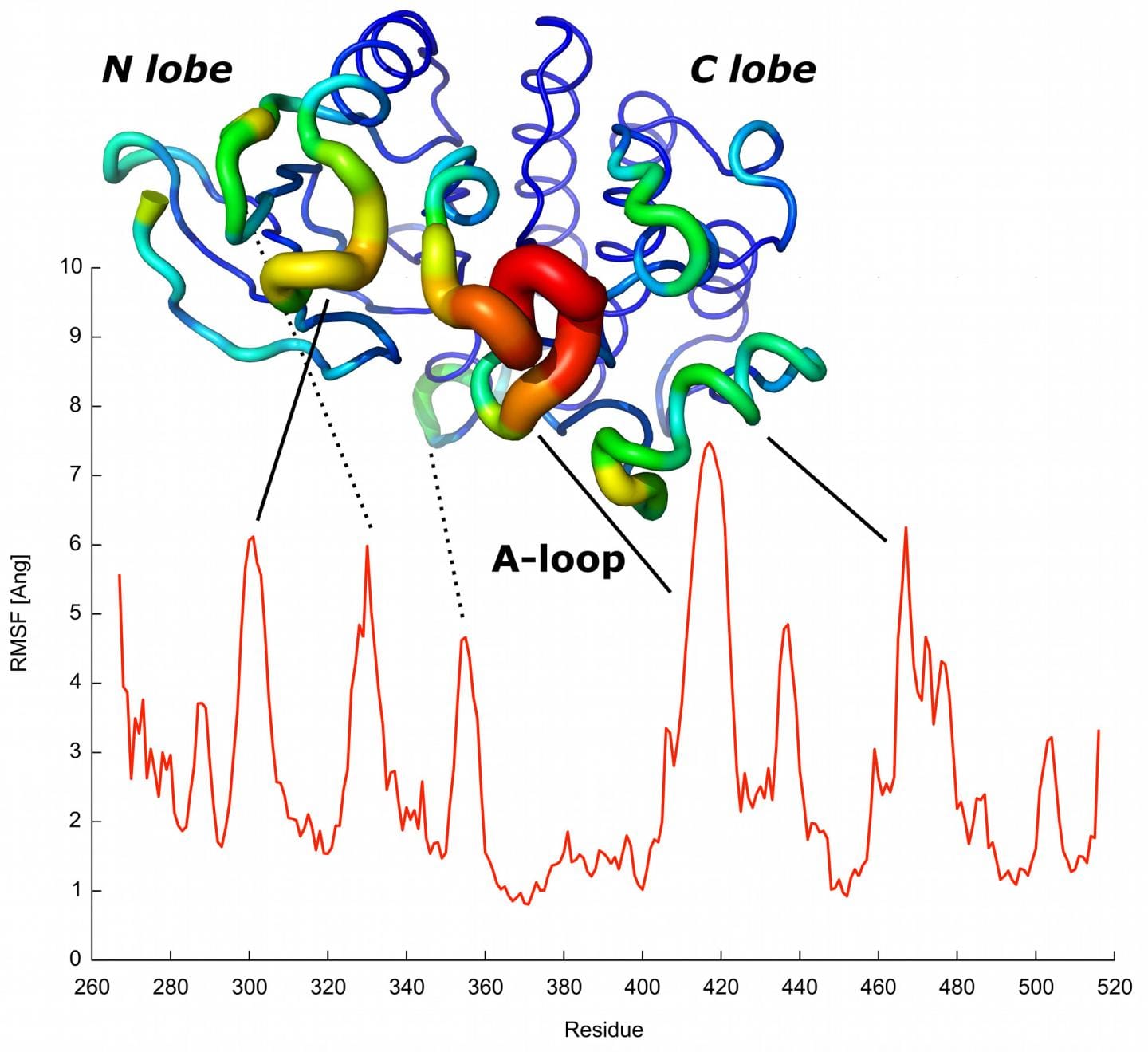
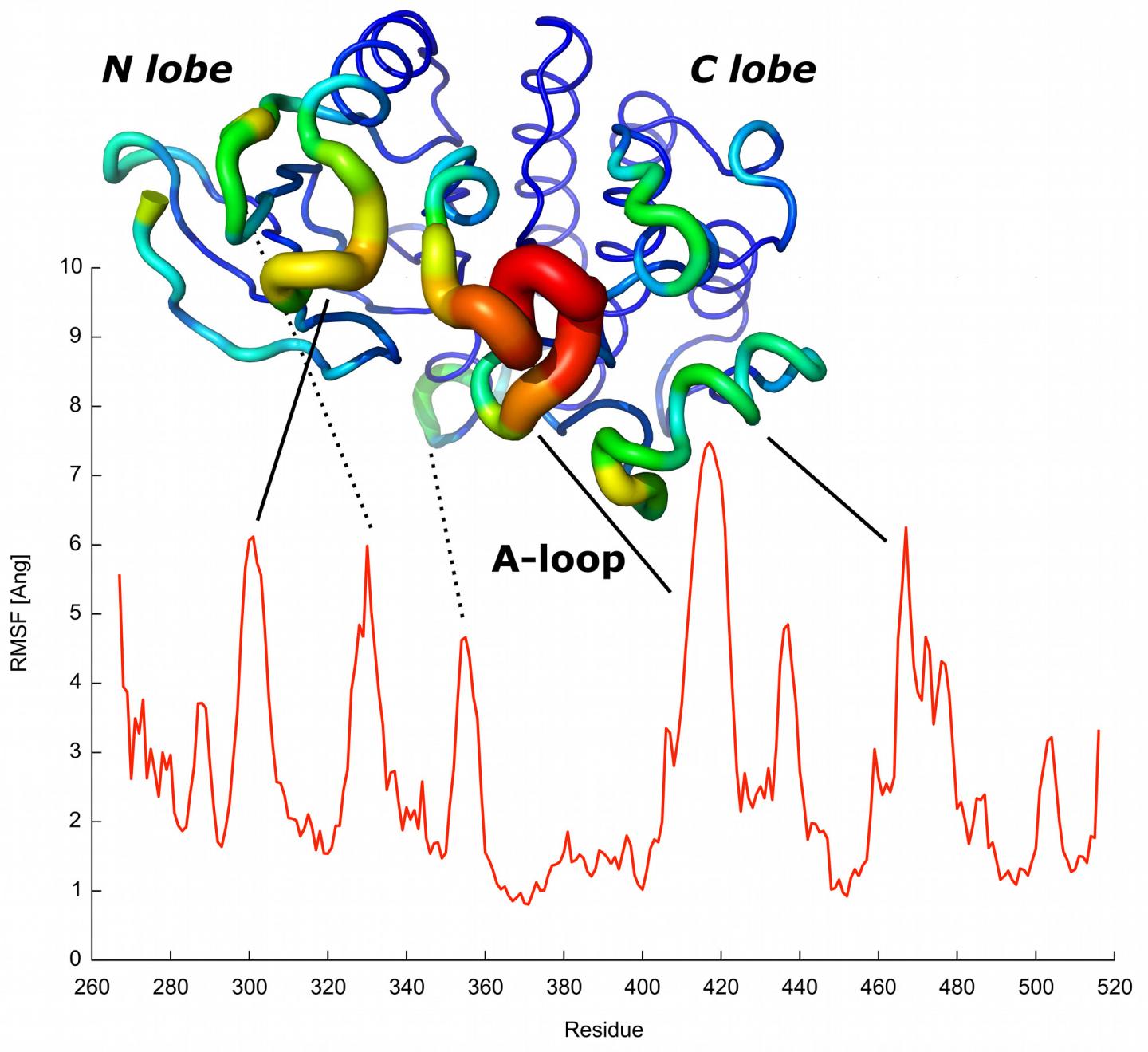
The red profile predicts the mobility of each protein residue. The most mobile residues predicted are indicated and matched with elements known to have a role in protein activation. Credit: CNIO.
Researchers from CNIO in Madrid and University College London have successfully predicted the movements a protein does to execute its functions. This remarkable advance in the protein dynamics field opens the door to a better drug design and new perspectives in the study of genetic diseases. The study has been published in the journal PNAS.
Proteins are biomolecules that carry out many cellular functions. They are formed by amino acids, small molecules that act as building blocks linked to one another to construct polymeric chains that will fold into a 3D structure. Structural biologists have developed computational methods that predict very accurately protein structure from just an amino acid sequence. This has been possible by analyzing the behavior of amino acid sequences in experimentally solved structures, mainly by X-ray crystallography and, to a minor extent, by nuclear magnetic resonance spectroscopy (NMR). Studying the sequences of proteins from the same family, scientists discovered that similar amino acid sequences fold in the same way, and that some couples of amino acids that are in contact in solved structures have co-evolved -have changed coordinately. All this information allows to anticipate what amino acids will be attracted to each other, and what final form the protein will adopt. The problem is that this final structure is not stable, as proteins need to move to fulfill their roles or to engage in interactions with other molecules. Protein dynamics haven´t been predictable so far.
Predicting movement from amino acids that evolved together
The research team developed a model that predicts protein dynamics by defining as mutually attractive those amino acids that have co-evolved, without taking into account any other information. With a prediction algorithm and a coarse-grained protein model, the researchers explored conformations that complied with the co-evolutionary norms. All the protein domains examined had a most representative structure that was coincident with the experimental one; and each segment of the protein domain showed variable mobility and alternative conformations, accountable for the protein motion range. The team concluded that protein dynamics can be predicted from just the amino acid sequence.
Source: CNIO
Reachout to Researchers with Our Extensive Marketing Network
Modern scientific marketing partner built for life science brands